
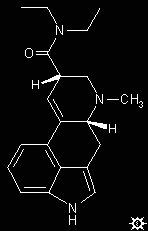
#26. LSD-25
|
| [3D .mol Image] |
![[3D .mol Image]](http://www.erowid.org/entheogens/lsd/images/archive/lsd_3d_mid.jpg){kind=link}
A suspension of 3.15 g d-lysergic acid hydrate and 7.1 g of diethylamine in 150 mL CHCl3 was brought to reflux with stirring. With the external heating removed, there was added 3.4 g POCl3 over the course of 2 min, at a rate sufficient to maintain refluxing conditions. The mixture was held at reflux for an additional 5 min, at which point everything had gone into solution. After returning to room temperature, the solution was added to 200 mL of 1 N NH4OH. The phases were separated, the organic phase dried over anhydrous MgSO4, filtered, and the solvent removed under vacuum. The residue was chromatographed over alumina with elution employing a 3:1 C6H6/CHCl3 mixture, and the collected fraction stripped of solvent under hard vacuum to a constant weight. This free-base solid can be recrystallized from benzene to give white crystals with a melting point of 87-92 °C. IR (in cm-1): 750, 776, 850, 937 and 996, with the carbonyl at 1631. The mass spectrum of the free base has a strong parent peak at mass 323, with sizable fragments at masses of 181, 196, 207 and 221.
This base was dissolved in warm, dry MeOH, using 4 mL per g of product. There was then added dry d-tartaric acid (0.232 g per g of LSD base), and the clear warm solution treated with Et2O dropwise until the cloudiness did not dispel on continued stirring. This opaqueness set to a fine crystalline suspension (this is achieved more quickly with seeding) and the solution allowed to crystallize overnight in the refrigerator. Ambient light should be severely restricted during these procedures. The product was removed by filtration, washed sparingly with cold methanol, with a cold 1:1 MeOH/Et2O mixture, and then dried to constant weight. The white crystalline product was lysergic acid diethylamide tartrate with two molecules of methanol of crystallization, with a mp of about 200 °C with decomposition, and weighed 3.11 g (66%). Repeated recrystallizations from methanol produced a product that became progressively less soluble, and eventually virtually insoluble, as the purity increased. A totally pure salt, when dry and when shaken in the dark, will emit small flashes of white light.
DOSAGE : 60 to 200 micrograms, orally
DURATION : 8 - 12 hrs
QUALITATIVE COMMENTS : In the case of LSD, it seems presumptuous to attempt to select typical comments for quotation. Literally thousands of reports are in the literature, from early exploratory research, to clinical applications for treatment of autism, of alcoholism, or mental illness, to assisting in psychotherapy and in the dying process, to the adventures of the military in both intelligence and chemical warfare, to innumerable anecdotal tales of pleasure and pain. Dozens of books have been devoted to these topics.
EXTENSIONS AND COMMENTARY : LSD is an unusually fragile molecule and some comments are in order as to its stability and storage. As a salt, in water, cold, and free from air and light exposure, it is stable indefinitely. There are two sensitive aspects of its structure. The position of the carboxamide attachment, the 8-position, is affected by basic, or high pH, conditions. Through a process called epimerization, this position can scramble, producing isolysergic acid diethylamide, or iso-LSD. This product is biologically inactive, and represents a loss of a proportionate amount of active product. A second and separate point of instability is the double bond that lies between this 8-position and the aromatic ring. Water or alcohol can add to this site, especially in the presence of light (sunlight with its ultraviolet energy is notoriously bad) to form a product that has been called lumi-LSD, which is totally inactive in man. Oh yes, and often overlooked, there may be only an infinitesimal amount of chlorine in treated tap water, but then there is only an infinitesimal amount of LSD in a typical LSD solution. And since chlorine will destroy LSD on contact, the dissolving of LSD in tap water is not appropriate.
There are many synthetic methods developed and reported for the preparation of LSD. All of them start with lysergic acid, and for that reason it has been listed as a Schedule III controlled drug, as a depressant, under Federal law. The amide lysergamide, a component of several varieties of morning glory seed, is also a controlled drug and, by law, a depressant. The earliest syntheses of LSD involved the used of an azide intermediate (the original Hofmann process, 1955), mixed anhydrides with trifluoroacetic anhydride (1956) or sulfuric anhydride (SO3-DMF on the lithium salt, 1959), with the peptide condensation agent N,N'-carbonyldiimidazole (1960), or with the acid chloride as the active intermediate with POCl3, PCl5 or thionyl chloride (1963) or just phosphorus oxychloride (1973). Most methods are faulted due to excessive moisture sensitivity, generation of side-products, or epimerization or inversion at the 8-position carbon to form d-iso-LSD. The POCl3 procedure is clean and fast, and is the preferred process today for the synthesis of a wide variety of substituted lysergamides.
The term LSD comes from the initials of the German for lysergic acid diethylamide, or Lysers�ure Diethylamid. The number "25" following it has many myths attached to it, such as it was the 25th form of LSD that Hofmann tried, or it was his 25th attempt to make LSD. From my own experience with chemical companies that are allied with pharmaceutical houses, I had assumed that the chemical name (which might be a mouthful for the pharmacologist) was simply replaced with a pronounceable code number equivalent. But the answer here is yet simpler. Hofmann, in his LSD, My Problem Child wrote: "In 1938, I produced the twenty fifth substance in a series of lysergic acid derivatives: lysergic acid diethylamide, abbreviated LSD-25 ... for laboratory usage."
Within a few years of the discovery of the extraordinary potency of LSD, a large number of close analogues were synthesized by Hofmann and his allies at Sandoz. Over the following decade many were tested in humans, both in patients and healthy subjects, with the qualitative descriptions and dosages published in the medical literature.
ALD-52. 1-Acetyl-N,N-diethyllysergamide. This material has been explored in the 50-175 microgram range and there are a number of human trials reported, with varying conclusions. One found that there was less visual distortion than with LSD and it seems to produce less anxiety and was somewhat less potent than LSD. Another report claimed it was more effective in increasing blood pressure. Yet another could not tell them apart. ALD-52 just may have been the drug that was sold as "Orange Sunshine" during the "Summer of Love" in the late '60's. Or "Orange Sunshine" may have been, really, LSD. This was the focus of a fascinating trial where two defendants were accused of distributing LSD, whereas they claimed that it was ALD-52 which was not an illegal drug. The prosecution claimed that as it hydrolyses readily to LSD, for all intents and purposes it was LSD, and anyway, you had to go through the illegal LSD to get to ALD-52 by any of the known chemical syntheses. The defendants were found guilty. And yet, I do not know who has actually measured the speed or ease of that reaction. If ALD-52 hydrolyses so easily to LSD, and the body is indeed a hydrolytic instrument, then these two drugs should be absolutely equivalent in every particular, This is the ergot equivalent of the psilocybin to psilocin argument, except this is an acetamide rather than a phosphate ester.
--indole-ring substituent-- at N-1 at C-2 Code -H
-COCH3
-CH3
-CH2OH
-CH2N(CH3)2
-H
-H
-CH3
-CH3-H
-H
-H
-H
-H
-B
-I
-Br
-ILSD-25
ALD-52
MLD-41
BOL-148
MBL-61
MIL
MLD-41. 1-Methyl-N,N-diethyllysergamide. The 1-methyl homologue of LSD is has more of somatic than sensory effect, has fewer visuals and is less well accepted than LSD, with the range of dosages being from 100 to 300 micrograms. This indicates that it is perhaps a third the potency of LSD which is in accord with both pupilary dilation and reflex action. However, the cardiovascular responses are actually increased. Besides being less potent than LSD, it appears to have a slower onset but it is equally long lived. There is cross-tolerance between MLD-41 and LSD.
BOL-148. 2-Bromo-N,N-diethyllysergamide. This synthetic ergot derivative, along with its 1-methyl homologue MBL-61 (mentioned below) should be used as powerful tools for studying the mechanism of action of LSD in the human animal. It does not have LSD-like effects in man. At 6 to 10 milligrams orally, there are some mental changes noted. But in another study, 20 milligrams was administered a day to a subject for 7 days, and there were no reported effects. And yet it is as potent a serotonin agonist as is LSD. How can serotonin be argued as a neurotransmitter that is a major player in explaining the action of psychedelic drugs, when this compound is nearly without activity.
There are some suggestions that an intervenous route may be more effective. I have heard of effects being noted at maybe a milligram and a short (2-3 hour) intoxicaion following 20 milligrams administered over a 20 minute period. I was involved many years ago in a study of radio-labelled BOL-148 which was made by the bromination of LSD. I was quite sure that the only radioactive material present was BOL-148, but there could well have been some unreacted LSD still present which would, of course, still be psychoactive. The synthesis is not clean -- I was tempted to make an entry for this compound if only to reproduce Albert Hofmann's original published experimental procedure. He reacted 13.2 grams of N-bromosuccinimide (in 400 mL dioxane, with 1.2 liters of dioxane containing 25 grams of LSD. This gave 11 grams of crude produce which had to be recrystallized. The radioactive syntheses uses effectively elemental bromine, and gave yields of from 5 to 15%. Visualize that reaction! A warm flask containing over a quart of warm solvent in which there was maybe half a million doses of LSD.
1-Hydroxymethyl-LSD, 1-dimethylaminomethyl-LSD and 2-iodo-LSD. These three additional compounds are shown here because they were described in a synthetic flurry that followed the discovery the activity of LSD. But at the moment I know neither their internal Sandoz codes nor if they had ever been explored in man. This is a kind of frustrating catch-all entry, in that the long index will send you here, and once here you realize that nothing is known. Well, at least the compounds are known, and perhaps there is something in the Sandoz vaults that might be interesting. I do not have access to them.
MBL-61. 2-Bromo-N,N-diethyl-1-methyllysergamide. This is the compound BOL-148 (mentioned above) with a methyl group attached to the 1-position of the indole ring (LSD has a hydrogen there). This wold be an even more tantalizing challenge to the serotonin theory for centrqal activity of the psuchedelics, in that it is without any activity in man at an oral dose of 14 milligrams (similar to the inactivity of the BOL-61 compound, but it is spome five times more potent as a serotonin agonist. With it, as with the iodiniated analogue MIL, there are many examples of the compromising of scientific integrity in the quest for funds and recognition. Both compounds are as effective as LSD itself in displacing labelled LSD that is bound to the post-synaptic serotonin receptor sites in animal brains. But neither of them show any LSD-like activity. But both have been labelled with 11-C or 122-I to give positrol emitting forms that can be administered to man and localized in a positrom emition tomographt instrument (a PET scanner).
I was at a meeting of a NIDA study section a few years ago, where some one presented some findings with a group of subjects who were complaining of continuing mental problems alledgedly due to LSD exposure. A chart was put up showing the outline of the brain showing the locations of the EEG foci that were observed in one of these subjects. Along side it was a PET scan showing the distribution of radioactive LSD in a subject. The purpose was to discuss the similarities and differences of the coordinates of electrical activity and radio-isotope concentration. I innocently asked what positron isotope had been used, as I did not know of any successful positron labelling of LSD. Carbon 11, I was told. Where in the molecule was the label incorporated, I asked. In the 1-position methyl group. It was finally acknowledged that the compound that had actually been used was 2-iodo-1-methyl-LSD, our MIL compound, which is quite a different world. A pharmacologist might say that they are similar in action (looking at serotonin, not psychedelic action), and achemist might say they are of similar structure (looking at the upper 80% of the moledule. But they are different compounds. This is a most subtle form of deceit. It is, in fact, out and out dishonest, but it looks good up there on the screen at a lecture.
Let me mention in passing, that there are three stereoisomers possible for d-LSD. There are d-iso-LSD, l-LSD, and l-iso-LSD. The inversion of the stereochemistry of the attached diethylcarboxyamido group of d-LSD gives the diastereoisomer (d-iso-LSD) which is a frequent synthetic impurity of d-LSD itself. The corresponding optical antipodes l-LSD and l-iso-LSD are also known and have been tasted. All three are completely inactive: d-iso-LSD shows no psychological changes at an oral dose of 4 milligrams; l-LSD none at up to 10 milligrams orally; and l-iso-LSD none at 500 micrograms orally. These dramatic decreases in potency show both the stereoselectivity of the native LSD molecule in producing its central effects, and the LSD-free purity of these isomers.
| indole | --- amide nitrogen substituents --- | ||
| R= | R= | R= | code name |
| -H -H -H -H -H -H -H -H -H -H -H -H -H -H -H -H -H -H -H -H -H -H -H -H -H -H -H -H -H -H -CH3 -CH3 -COCH3 -CH3 |
-H -CH3 -CH2CH3 -(CH2)2CH3 -CH(CH3)CH2OH -CH(CH3)CH2OH -(CH2)CH3 -CH(CH3)CH2CH2OH -(CH2)4CH3 -CH(CH3)CH2CH2CH3 -CH(CH2CH3)2 -(CH2)5CH3 -CH(CH3)CH2CH2CH2CH3 -(CH2)6CH3 -CH(CH3)CH2CH2CH2CH2CH3 -CH3 -CH2CH3 -(CH2)2CH3 -CH(CH3)2 -CH(CH3)CH2C6H5 -CH2CH3 -(CH2)2CH3 -(CH2)2CH3 -CH(CH3)2 -CH2CH=CH2 -(CH2)2CH3 -CH2CH=CHCH2- -CH2CH2CH2CH2CH2- -CH2CH2OCH2CH2- -CH(CH2CH3)CH2OH -COCH3 |
-H -H -H -H -H* -H -H* -H* -H -H* -H -H -H* -H -H* -CH3 -CH3 -CH3 -CH3 -CH3* -CH2CH3 -CH2CH3 -(CH2)2CH3 -CH(CH3)2 -CH2CH=CH2 -(CH2)3CH3 -H -H* -CH2CH3 |
LA-111 LAE-32 Ergonovine Methergine DAM-57 LAMP LSD-25 DAL LPD-824 LSM-775 MLA-74 UML-491 APA-10 MPD75 |
In the amides marked with "*" there has been the introduction of a new asymmetric center, which of course doubles the number of isomers that is possible. In each case the resulting two optical forms were prepared separately, and evaluated separately as to their pharmacology.
This listing is not intended to be thorough, but it is shown to suggest the amount of synthetic effort that has been made towards the exploring and understanding the high potency associated with those two remarkably important ethyl groups on the amide nitrogen of LSD. I have given the Sandoz code names, again, as far as I know them. Although none of these really warrant a dedicated recipe, there is sufficient animal and human pharmacology reported to justify listing them below as separate items. Most of these reports appeared in the mid-1950's but some studies are still being done and papers are published even today with new ideas but, sadly, only with animal pharmacology. I have been as guilty as the next person who has tried to mount all these compounds into a table with a "human potency" factor that compares them directly to LSD. This is an uncomfortable simplification. Here are the actual reported observations, and I'll let the reader provide his own potency index.
LA-111, ergine, d-lysergamide. This is an active compound and has been established as a major component in morning glory seeds. It was assayed for human activity, by Albert Hofmann in self-trials back in 1947, well before this was known to be a natural compound. An i.m. administration of a 500 microgram dose led to a tired, dreamy state with an inability to maintain clear thoughts. After a short period of sleep, the effects were gone and normal baseline was recovered within five hours. Other observers have confirmed this clouding of consciousness leading to sleep. The epimer, inverted at C-8, is isoergine or d-isolysergamide, and is also a component of morning glory seeds. Hofmann tried a 2 milligram dose of this amide, and as with ergine, he experienced nothing but tiredness, apathy, and a feeling of emptiness. Both compounds are probably correctly dismissed as not being a contributor to the action of these seeds. It is important to note that ergine, as well as lysergic acid itself, is listed as a Schedule III drug in the Controlled Substances Act, as a depressant. This is, in all probability, a stratagem to control them as logical precursors to LSD.
LAE-32, N-ethyllysergamide. Different people have observed and reported different effects, with different routes of administration. Subcutaneous administrations of from 500 to 750 micrograms have been said to produce a state of apathy and sedation. Clinical studies with dosages of 500 micrograms i.m. were felt to be less effective than the control use of 100 micrograms of LSD. And yet, oral doses of twice this amount, 1.6 milligrams, have been said to produce a short-lived LSD-like effect with none of these negatives.
LPD-824, N-Pyrrolidyllysergamide. Five trials at a dosage of 800 micrograms orally led to the reporting of a fleeting effect that was similar to one tenth this amount of LSD.
LSM-775, N-Morpholinyllysergamide. There are conflicting reports; one states that 75 micrograms is an effective dose, comparable to a similar dose of LSD, and the other stated that between 350 and 700 micrograms was needed to elicit this response, and that there were fewer signs of cardiovascular stimulation and peripheral toxicity.
DAM-57, N,N-Dimethyllysergamide. This compound did induce autonomic disturbances at oral levels of some ten times the dosage required for LSD, presumably in the high hundreds of micrograms. There is some disagreement as to whether there were psychic changes observed.
DAL, N,N-Diallyllysergamide. As the tartrate salt, there is at best a touch of sparkle seen at 600 micrograms orally, but there is a sedation also reported. It is certainly an order of magnitude less potent than LSD itself.
UML-491, Methysergide, Sansert. This is the synthetic homologue of methergine (1-methyl) and is employed clinically as a treatment for migraine headaches. When the usual therapeutic dosage of two milligrams is scaled up by a factor of ten, there is a profound LSD-like response described by most subjects. A number of these ergot analogues from nature can be considered as potential precursors for the preparation of LSD. But here, there is a 1-methyl group that is effectively permanently attached, so it cannot play this role.
ands a separate entry.
Ergonovine is a naturally occurring, water-soluble ergot alkaloid, found in both ergot preparations and in many species of morning glory seeds, and there are several reports of LSD-like action at oral levels of between two and ten milligrams. It has an important use in obstetrics, again as an oxytocic, at about a tenth of this dose. This pharmacological potential must be respected in psychopharmacological trials. The one-carbon homologue (the butanolamide rather than the propanolamide) is called methergine or methylergonovine. It is a synthetic ally and is orally effective as an oxytocic at a dosage of 200 micrograms. It also has an LSD-like action at ten times this level.
Although there are many other chemical treasures in the ergot fungal world, I would like to wrap this commentary up with a return to the topic of morning glory seeds. Four additional alkaloids of the ergot world must be acknowledged as being potentially participating factors in the MGS story. With each of these, the primary ergoline ring system is largely intact but the amide function is completely gone. The carboxyl group has been reduced to the alcohol to give elymoclavine. There is the related molecule present which is the isomer with the double bond moved to be conjugated with the aromatic ring; it is called lysergol. There is the same molecule but with a hydroxy group attached to the 8-position carbon atom (an ethyleneglycol!) ; it is called penniclavine. And lastly, that D-ring can actually be opened between the 5 and 6 positions, to give us a secondary amine tryptamine derivative, chanoclavine. To be completely anally retentive in this Ipomoea inventory, mention must be made of five alkaloids that are present in truly trace amounts, all of which have no oxygen atoms present whatsoever on that substitution on the ergoline 8-position. These are the 8-methyl isomers agroclavine, setoclavine, festuclavine and cycloclavine, and the methylene analogue lysergene. These structures in effect define absolute obscurity, and most probably do not contribute to the morning glory intoxication state. But the others, some present is sizable amounts, may someday help explain why the pharmacology of these seeds is so different than that of the major isolates, the ergines.