
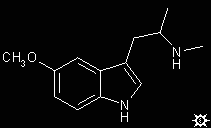
#55. a,N,O-TMS
TRYPTAMINE, 5-METHOXY-a,N-DIMETHYL; INDOLE, 5-METHOXY-3-[2-(METHYLAMINO)PROPYL]; 5-METHOXY-a,N-DIMETHYLTRYPTAMINE; 5-METHOXY-3-[2-(METHYLAMINO)PROPYL]INDOLE; a,N,O-TRIMETHYLSEROTONIN; SEROTONIN, a,N,O-TRIMETHYL
|
| [3D .mol Image] |
A suspension of 1.7 g electrolytic iron dust in 10 mL of 80% aqueous acetic acid was heated on the steam bath until there were clear signs of hydrogen evolution. To this stirred suspension there was added 0.50 g 5-methoxy-3-(2-nitropropenyl)indole a bit at a time, over the course of 2 min. The heating and stirring was continued for 30 min by which time TLC analysis (CH2Cl2/hexane, silica) showed the starting material to be gone, and there were two new spots, one slower moving and one at the origin. The reaction mixture was poured into 100 mL of a H2O/CH2Cl2 mixture, and filtered through paper. The two phases were separated, and the aqueous phases extracted with an additional 2x50 mL CH2Cl2. The pooled organic extracts were washed once with saturated aqueous K2CO3, and the solvent removed under vacuum. The resulting residue was distilled at 0.08 mm/Hg to give 5-methoxyindol-3-yl acetone as a colorless oil which came over at 215-230 °C. The product weighed 0.24 g, had a carbonyl absorption at 1710 cm-1, and had an acceptable fragmentation pattern by GCMS.
To 20 mL methanol, there was added 1.17 g of 5-methoxyindol-3-yl acetone, 4.3 g CH3NH2 hydrochloride, 0.5 g NaCNBH3 and sufficient concentrated HCl/MeOH to bring the pH down to a yellow color on damped, broad range pH paper. The reaction was stirred at room temperature, with periodic addition of more acid as needed, over the course of several days. The reaction mixture was poured into dilute sulfuric acid, washed twice with CH2Cl2, made basic with dilute NaOH, and extracted with 3x50 mL CH2Cl2. After the removal of the solvent under vacuum, the residue (0.76 g) was distilled at 180-190 °C at 0.05 mm/Hg to give 0.65 g a,N,O-trimethylserotonin (a,N,O-TMS) as a colorless oil. It did not crystallize, nor were any solid salts of it obtained. MS (in m/z): C3H8N+ 58 (100%); methoxyindolemethylene+ 161/160 (19, 7%); parent ion 218 (<1%).
DOSAGE : 10 - 20 mg, orally
DURATION : 6 - 8 hrs
QUALITATIVE COMMENTS : (with 16 mg, orally) "It was maybe a plus two, but there wasn't really much of anything. My body felt safe, was safe. The only strong negative, negative only to me, was the nature of my dreaming that night; shallow, a faint metallic flavor, a distinct lack of depth or dimension. A waste of good dreaming time."
(with 16 mg, orally) "I got as far as this would go, at about an hour and a quarter. There was nothing in the visual field, but I was unquestionably somewhere. I was quite horny, and the erotic was both excellent and satisfying. Tried writing, and it seemed easy. There was no fantasy, no color enhancement, and not much in the way of eye-dilation or appetite loss. Nothing much anywhere at all. As with Oakland, no there, there. And by the seventh hour, there was baseline, with a residual feeling of having been cleansed."
(with 20 mg, orally) "I was in quite a depressed state, but I don't think it was the a,N,O. But it certainly didn't lift the depression. Everything I saw confirmed my growing despair about the human race and I concluded, after a few hours, that we have doomed ourselves. I am tired at being angry at it all. Enough already."
EXTENSIONS AND COMMENTARY : There is a sadness felt with most of the published efforts to form sweeping correlations between the structure of a molecule and its biological activity. This relationship is called a SAR, or a Structure Activity Relationship, and there are journals that are dedicated to just this form of analysis.
One needs a large collection of compounds of known structure, and all of them must be of known pharmacological activity. And one needs a computer of some sort. One considers all aspects of the structure such as bond energies, electronic charge densities, molecular lengths, widths and thicknesses, degrees of freedom or of constraint, anything that can be calculated or measured. Then one assigns an independent variable coefficient to everything, constructs some additive equation where these coefficients equal something else, and then compares that something else to the biological activity. Push the "go" button on the computer, and let everything be varied clear across the map, until the calculated solution of the equation makes the best match with the value of pharmacological activity. Then one has a SAR with a statistical measure of goodness of fit, and it then can be used to predict the activity of new structures, which are yet untried, pharmacologically.
And there is the essence of why this entire process is ineffective. Prediction is the heart of this procedure, and prediction is never brought to bear. Let us take a new structure that is not in the original collection of structures, and let us make a prediction as to its, let us say, psychedelic potency. But no one ever tries it out for any of a number of reasons. Maybe the new compound is never synthesized. Or maybe it is synthesized, but never evaluated pharmacologically. The synthesist does not care, or is uninterested, or is restrained by the legal complications that might ensue. Or he does explore it, but chooses not to publish. Almost never is a prediction tested. What is more likely to happen, is that a new input of biological activity and structure variation is uncovered (for which there is no published prediction) and this data is tossed into the mill, and a new set of "more valid" coefficients is calculated, and the SAR becomes touted as a more accurate predictor. But, always remember, that without prediction and challenge, there is no inventive value from the SAR game. It simply organizes what is known, but creates nothing new.
This is a role that I would have loved to see a,N,O-TMS play. At the time of its first synthesis its biological activity was, by definition, completely unknown. Let's cast its shadow up against the structures that were known, and with known activity. What would you predict? The most logical archetype to use as a starting point is the primary amine homologue, a,O-DMS. This is an extremely potent, quite long-lived tryptamine that still ranks up there as the most potent, or nearly so, of all the simple substituted tryptamines. It is orally active. It lasts for many hours. It is completely wild as to visual distortions and illusions. It consistently leads to dramatic, perhaps frightening, but certainly memorable dreams. Three or four milligrams are unmistakably adequate. I would have loved to have had an SAR jock predict what changes would come from the simple addition of an N-methyl group. No one out there predicted this for me, and I have now completely abandoned the art of prediction, at least via the SAR technique. My motto is, make 'em, and taste 'em.
To base structures that are stimulants (amphetamine, for example) an added N-methyl group enhances potency and richness. With MDA, for example, one gets MDMA, not more potent, but of an entirely different form of psychological magic. However, with all the other explored primary amine phenethylamine psychedelics, the potency and the quality of action are effectively lost. With tryptamines, however, the N-methyl groups appear to be needed for full, robust activity. Here, the loss of an N-methyl group might well detract from full potency, and the final unmethylated product (DMT becoming simply tryptamine) will be relatively weak and uninteresting. If a,N,O-TMS had been active at one milligram, then the MDMA explanation is obviously correct. If a,N,O-TMS had been active only at a meager level of twenty milligrams, then the DMT explanation would appear to be correct. It is much less active. It is not spectacular. All you SAR scientists, take this new data, toss it into the maws of computer calculation, and come out with better coefficients.
With this, now, as a challenge, predict for me the potency of a,N,N,O-tetramethylserotonin. Here is a compound that has not been yet synthesized, but which carries the second N-methyl group (yet closer to DMT at the nitrogen atom and probably more potent) and yet a structural kiss of death (as to potency) in the MDA/MDMA world. Will it be up? Will it be down? I am afraid that the "make 'em and taste 'em" procedure is the only one that I can trust.
Good luck.