|

|
|
|
|---|---|
|
|
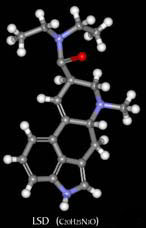
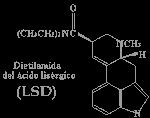
Preparatorios preliminares: El material de partida debe ser algún derivado del ácido lisérgico, del ergot, del grano del centeno, de las semillas de (morning glory), o bien obtenido por alguna vía sintética. Las preparaciones #2 y #3 deben partir de ácido lisérgico solamente, el cual se prepara a partir de su amida como se describe a continuación: 10g de alguna amida de ácido lisérgico obtenida por vía natural se disuelve en 200ml de una disolución de metanol+KOH, y el metanol se elimina inmediatamente al vacio. El residuo es tratado con 200ml de una disolución acuosa de KOH del 8%, y la mezcla es calentada en bańo de vapor durante una hora. Se hace pasar una corriente de nitrógeno en el frasco durante el calentado y el NH3 gas producido se valora con HCl para seguir el proceso. La disolución alcalina debemos neutralizarla con ácido tartárico y como indicador usaremos rojo congo. A continuación la disolución acuosa evaporada se filtra y se limpia por extracción con éter. Se le hace una digestión con metanol para eliminar algunos de los materiales coloreados de los cristales de ácido lisérgico. Preparamos la luminosidad del laboratorio para que sea similar a una habitación oscura. Debemos tener preparadas luces rojas y amarillas, debido a que los derivados del ácido lisérgico se descomponen en presencia de la luz natural. Debemos usar guantes de goma debido a la naturaleza altamente venenosa de los alcaloides del ergot. Un secador de pelo, o mejor, un evaporador es necesario para acelerar algunos pasos en los que hay que evaporar. Preparación #1: Paso 1. Usar luz amarilla. Colocamos un volumen de alcaloide de ergot pulverizado en un frasco pequeńo y ańadimos dos volumenes de hidracina anhidra. Un procedimiento alternativo es usar un tubo sellado en el que se calientan los reactivos a 112şC. La mezcla se calienta a reflujo durante 30 minutos. Le ańadimos 1.5 volumenes de agua y hervimos 15 minutos. Al introducir la disolución en el frigorifico cristalizará la hidracina del ácido isolisergico. Paso 2. Usar luz roja. Enfriamos todos los reactivos y debemos tener hielo cerca para usar. Disolvemos 2.82g de hidracina rapidamente en 100ml de una disolucion 0.1N de HCl que previamente habremos enfriado con hielo. Usaremos un bańo de hielo para mantener el vaso en el que llevamos a cabo la reaccion a 0şC. 100ml de una disolución 0.1N de NaNO2, enfriada con hielo, se ańade a la mezcla y después de 2 o 3 minutos de agitación vigorosa se ańaden 130ml mas de HCl y se agita otra vez en bańo de hielo. Después de 5 minutos de agitación se neutraliza la solución con una disolución saturada de bicarbonato sódico y realizamos una extracción con éter. Eliminamos la disolución acuosa y tratamos de disolver la sustancia viscosa en éter. Ajustamos la disolución de éter por adicción de 3g de dietilamina por cada 300ml de éter extraido. Lo dejamos en ausencia de luz en una habitación que calentamos gradualmente hasta 20şC durante 24 horas. Evaporamos al vacío y tratamos como se indica en la sección de purificación para la conversión de amidas de ácido iso-lisérgico en amidas de ácido lisérgico. Preparación #2: Paso 1.Usar luz amarilla. 5.36g de d-ácido lisérgico se suspende en 125ml de acetonitrilo y la suspensión se enfría a unos -20şC en un bańo de acetona enfriada con hielo seco. A la suspensión se le ańade una disolución fría (a -20şC) de 8.82g de anhídrido trifluoroacético disueltos en 75ml de acetonitrilo. Se deja la mezcla a -20şC durante hora y media. En este tiempo los materiales en suspensión se disuelven el d-ácido lisérgico se transforma en la mezcla de anhídrido lisergico y ácido trifluoroacético. El anhídrido puede ser separado en la forma de aceite por evaporación del disolvente al vacío a una temperatura de 0şC, pero esto no es necesario. De todas formas debe permanecer anhídro. Paso 2. Usar luz amarilla. La disolución de anhídro mezclado con acetonitrilo del paso 1 se ańade a 150ml de una segunda disolución de acetonitrilo que contiene 7.6g de dietilamina. La mezcla se mantiene en una habitación a temperatura ambiente durante 2 horas. El acetonitrilo se evapora al vacío dejando un residuo de LSD-25 más otras impurezas. El residuo se disuelve en 150ml de cloroformo y 20ml de agua-hielo. El cloroformo se elimina y se le realiza una extracción a la disolución acuosa con varias porciones de cloroformo. Las porciones de cloroformo se combinan para lavarlas con 4 porciones de 50ml de agua-hielo. La disolucion de cloroformo se seca sobre Na2SO4 y se evapora al vacio. Preparación #3: Este procedimiento tiene un buen rendimiento y es muy rápido también, origina una pequeńa cantidad de ácido iso-lisérgico (su efecto es medianamente desagradable). De cualquier forma, la estequiometría debe ser exacta o el rendimiento no sera óptimo. Paso 1. Usar luz blanca. SO3 es producido en estado anhídro por descomposición de sulfato férrico anhídro a una temperatura de 480şC. Mantenemos SO3 en condiciones anhídras. Paso 2. Usar luz blanca. Un recipiente de 22l encajado en un bańo de hielo, condensador, con embudo de llenado y agitador mecánico es llenado con 10 a 11 litros de dimetilformamida (destilada recientemente a presión reducida). El condensador y el embudo de llenado se protegen de la humedad atmosférica. 2 lb de SO3 se introducen con cuidado y se agita muy cautelosamente durante 4 o 5 horas (la adicion de SO3 debe ser poco a poco durante las 4 o 5 horas). La temperatura se mantiene en el intervalo 0-5şC durante la adición. Después de la adición se continua agitando durante 1 o 2 horas hasta que los cristales de sulfuro de trióxido-dimetilformamida se hayan disuelto formando un complejo. El reactivo se lleva a una pipeta automática cerrada aislada del aire para poner usar la cantidad deseada de reactivos, y los mantenemos frío. Aunque el reactivo, que es coloreado, puede cambiar de amarillo a rojo, lo podemos almacenar en frío durante 3 o 4 meses que permanecerá inalterado. Una licuota de reactivo se disuelve en agua y se valora con NaOH standart hasta el punto final de la fenolftaleína. Paso 3. Usar luz roja. Se disuelven de 7.15g de d-ácido lisérgico monohidratado (25mmol) y 1.06g de hidróxido de litio hidratado (25mmol) en 200ml de MeOH preparado. El disolvente es destilado en bańo de vapor a presión reducida. El residuo de lisergato de litio se disuelve en 400ml de dimetilformamida anhidra. De esta disolución se destilan unos 200ml de DMF anhidra a una presión de 15 ml de Hg a través de una columna de hélices de 12 pulgadas. La disolución resultante de lisergato de litio anhidro se enfría a 0şC, con agitación y se trata rápidamente con 500ml de la disolución de SO3-DMF (1.00 molar). La mezcla es agitada en frío durante 10 minutos y luego se ańaden 9.14g (125mmol) de DMF. El agitado y enfriado se prolongan durante 10 minutos más, tras los que se ańaden 400ml de agua. Después se ańaden 200ml de una disolución salina saturada. La amida que se produce es aislada por varias extracciones con porciones de 500ml de dicloroetileno. El combinado que se extrae es secado y luego concentrado hasta que parezca un jarabe a presión reducida. No se debe calentar el jarabe durante la concentración. El LSD cristalizara, pero los cristales y las aguas madres deben ser cromatografiadas de acuerdo con las instrucciones de purificación. Purificación del LSD: El material obtenido a partir de cualquiera de las tres preparaciones anteriores puede que contengan ácido lisérgico y amidas del ácido iso-lisérgico. La preparacion #1 contiene en su mayor parte dietilamida del ácido iso-lisérgico que debe ser transformada previa separación. Para esta sustancia, realiza primero el paso 2 de la purificación. Paso 1. Usar habitacion oscura con luz UV de larga longitud de onda. El material se disuelve en una mezcla 3:1 de benceno y cloroformo. Carga la columna de cromatografía con una gota de alumina básica en benceno. Una pulgada en la columna equivale a 6 pulgadas de longitud. Saca poco a poco el disolvente hasta el tope de la columna de alumina y ańade cuidadosamente una licuota de disolución de LSD formada por 50ml de disolvente y 1g de LSD. Hazlo correr por la columna seguido por el movimiento más rapido de la banda fluorescente. Después de que este haya sido cogido, limpia el material sobrante de la columna lavandola con MeOH. Usa la luz ultravioleta para prevenir excesivos dańos a los compuestos. Evapora la 2Ş fracción al vacío y ponla a un lado para el paso 2. La fracción que contiene LSD puro se concentra al vacío y el jarabe cristalizara lentamente. Este material se puede convertir en el tartato usando ácido tartárico y el tartato de LSD se cristaliza convenientemente. Su punto de fusión anda por los 190-196şC. Paso 2. Usar luz roja. Disuelve el residuo de haber limpiado la columna de cromatografía con metanol en una cantidad minima de alcohol. Ańade dos veces su volumen de una disolución alcohólica con KOH (4N) y deja que la mezcla permanezca a temperatura ambiente durante varias horas. Neutralizala con HCl diluido, hazla ligeramente básica con NH4OH y extrae el producto con cloroformo o dicloroetileno como en las preparaciones #1 y #2. Evapora al vacío y realiza la cromatografía como se indica en el paso 1 de la purificación. Nota: Los compuestos de ácido lisergico son inestables frente al calor, la luz y el oxígeno. Te puede ayudar a conservarlos ańadir ácido ascórbico como antioxidante, mantener el recipiente bien cerrado, recubierto de laminas de aluminio para evitar la luz y en el frigorifico. Enjoy It ! |